News & Articles
Introducing LEAP: (Machine) Learning from Evidence to Assess Pathogenicity
Carmen Lai
If you’re a human reading this post, your DNA is about 99.9% the same as mine. It’s the 0.1%, our genetic variations, which make each of us unique. One of our most important jobs at Color is interpreting the significance of each of our clients’ variants. It’s especially important that we accurately interpret pathogenic variants — those that are associated with increased disease risk. These can be life-changing results for our clients, so we’re constantly looking for ways to improve our practices and methods.
Today, we’re pleased to announce a fun achievement for one of our newest interpretive methods: Color recently won CAGI’s breast cancer variant prediction challenge, ahead of 10 other competing models.
If you’re new to genetics, you might wonder how scientists determine which variants are pathogenic. There are four to five million unique variants per human genome, most of which scientists have never studied or even seen. Most of these variants are benign and don’t pose an additional disease risk. Identifying pathogenic variants at this scale can be a huge challenge. To tackle it, Color, like other clinical laboratories, relies on a team of variant scientists aided by custom-designed software. Earlier this year, we worked with our scientists to develop LEAP (Learning from Evidence to Assess Pathogenicity), a machine-learned approach to determining whether a given genetic variant is associated with higher cancer risk. We recently published our approach and implementation details. Apart from winning the CAGI challenge, LEAP has been an invaluable internal tool for our variant scientists.
Why machine learning? Over the years, we’ve found that this technology can play a critical role in improving the efficiency and accuracy of our internal clinical and lab processes. This insight will be unsurprising to computer scientists, but we’re among the first clinical genetics labs to deploy machine learning in a variety of novel contexts. Apart from LEAP, we rely on machine learning to improve early detection of samples that are likely to fail when processed by our lab, and have developed novel variant confidence models to improve bioinformatic specificity and streamline secondary lab confirmations. We also recently deployed Google’s DeepVariant model as part of our growing ensemble of variant callers.
In this post, we’ll introduce variant interpretation, our thought process for applying and leveraging machine learning, various forms of model validation and pressure testing, applications in a clinical setting, and next steps we’d like to explore.
Background
Variant interpretation is the process of assigning variants to clinically relevant categories as recommended by ACMG based on the level of evidence. Variants can generally be classified as pathogenic (associated with increased disease risk), benign (not associated with increased disease risk), or of uncertain significance (VUS, not enough evidence at this time). The review process can be complex: variant scientists review multiple types of evidence for each case, some of which are structured (e.g., protein functional impact scores, population frequencies) and some unstructured (e.g., literature content, health histories).

This process shares many characteristics with other domains where machine learning is effective. There’s a database determined by experts (pathogenic vs. benign variant classification) and a large number of input signals with varying weights and importance that drive these expert decisions. What’s missing is a generalizable mapping between the two.
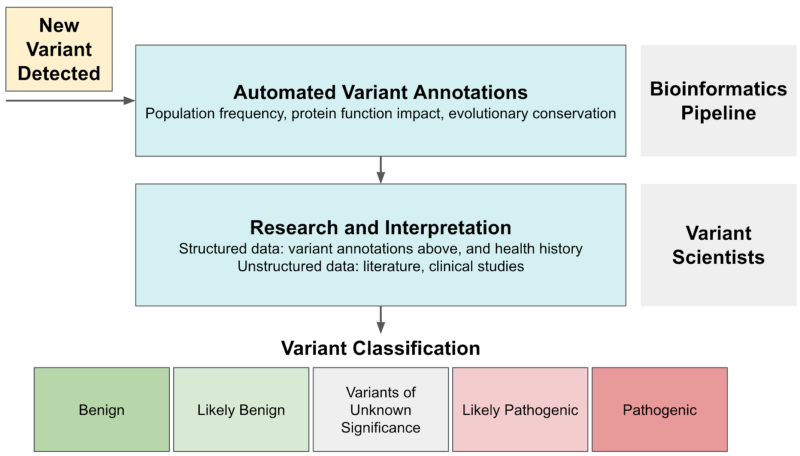
Our Data Science team saw an opportunity to use machine learning to build that map. Our primary goal in developing LEAP was to use Color’s knowledge base as a quality control mechanism for future classifications. Completely automating variant interpretation was not a goal for several reasons:
– The cost of even just one false positive or false negative is high
– Some forms of evidence (e.g., literature content) are difficult to encode computationally
– We want to augment existing systems and empower domain experts who know them best
Rules-Based Approach
We first built internal tooling to facilitate variant interpretation by aggregating many sources of evidence while recording classification updates. As an initial effort to reduce manual workload, we implemented rule-based classification criteria to allow automatic interpretation in cases that provide strong enough evidence. Common auto-classification rules include:
– Population frequency: commonly occurring variants detected in the broader population (>5%) or subpopulations (>2%) are benign.
– Variant location: variants located towards the end of designated transcripts (gene regions that are transcribed into RNA and then translated into a protein) are unlikely to have impact on protein transcription, and are therefore benign.
But strong assertions like these only apply to a small minority of variants. Most variants are rare, and rare variant interpretation is more challenging and time-consuming, due to limited and potentially conflicting information. To standardize the process across labs and institutions, the ACMG established a framework for variant interpretation, but this framework can become unreliable for more complex cases.
Machine-Learned Approach
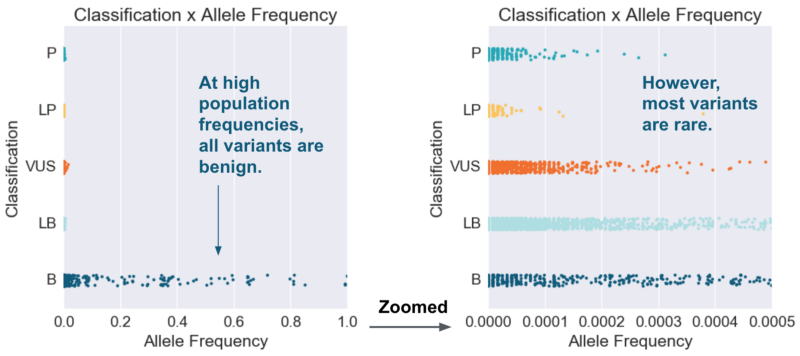
How might machine learning improve upon simple rules? Let’s start with one of our common auto-classification rules: commonly occurring variants (population frequency >5%) make a strong case for a benign classification. If we plot population frequency against variant classification, we see that this rule holds in the zoomed-out view, and that some benign cases can be separated from other classifications using a population frequency cutoff. As we zoom in, however, we see that most variants with different classifications are not easily separable based on population frequency alone.
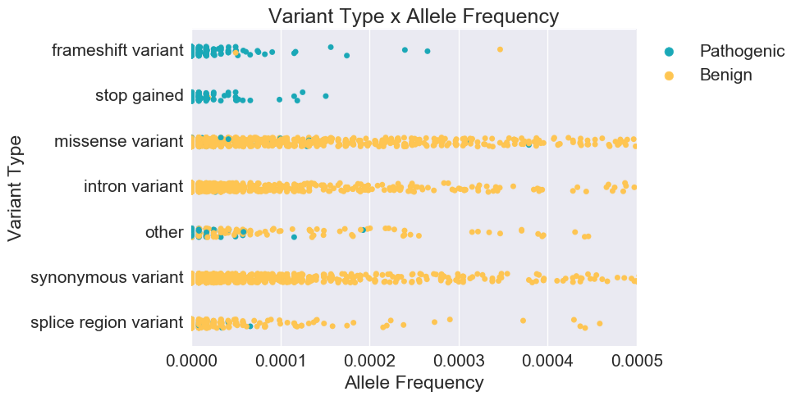
Variant scientists would then add that classification also depends on variant type. Plotting variant type alongside population frequency, we see clearer separation between pathogenic (P, LP) and benign (B, LB). In fact, while population frequency is known to be strong evidence for classifying some cases, we see below that for most cases, variant type is actually a stronger predictor of overall pathogenicity. Instead of relying on single-feature rules with arbitrarily determined cutoff values, we can use machine learning to generate rules and weight evidence based on the data itself.
Model Description and Validation
At a high level, LEAP was trained using L2-regularized logistic regression, and outputs a probability of pathogenicity and the underlying drivers of a given prediction. Our priority in model selection was explainability, and logistic regression not only gives us transparency into the individual weights and contribution of each feature, but also achieves decent performance. We experimented with other models as well, and found only marginal improvement in performance while explainability became limited.
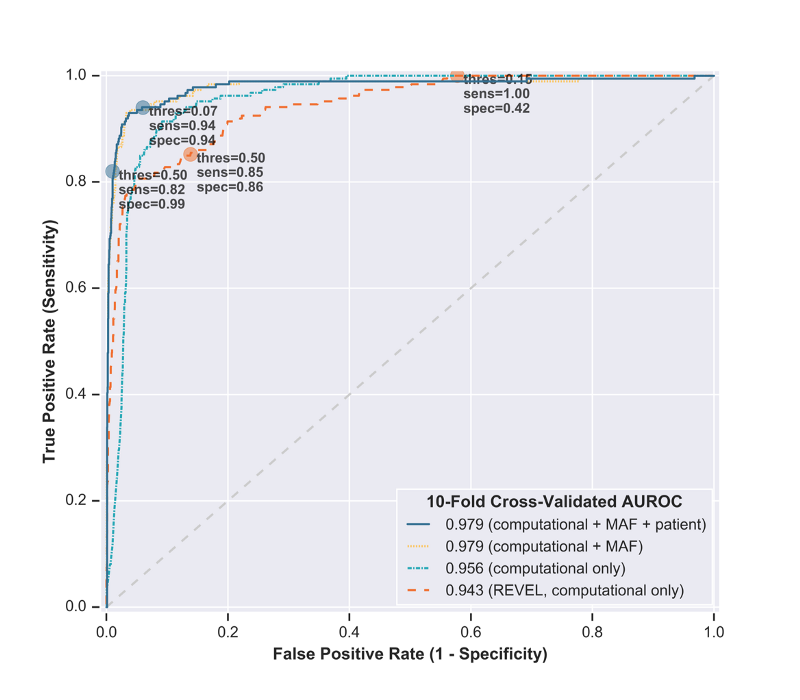
As internal validation of LEAP’s accuracy and robustness, we assessed predictions generated from two holdout methods:
– Standard: 10-fold cross-validation, where 90% of the dataset was used to train the model and predict on the other 10%.
– By Gene: gene holdout cross-validation, where all but one gene was used to train the model and predict on one held-out gene. This demonstrated LEAP’s extensibility to multiple cancer loss-of-function genes, even when the entire gene was absent from training.
Further validation would require seeing how LEAP performs on a holdout set outside Color, with difficult cases with which the model wasn’t trained. The CAGI challenge gave us this opportunity. The challenge involved variant interpretation in the BRCA1 and BRCA2 gene variants, which are most strongly associated with risk for breast and ovarian cancers. Variants used for assessment were mostly missense substitution variants, which are more challenging to interpret due to the unclear impact on protein synthesis and clinical function. Their model assessment was based on multi-class AUROC (area under the receiver operating characteristic) scores, which takes into account trade-off between false negatives and false positives.
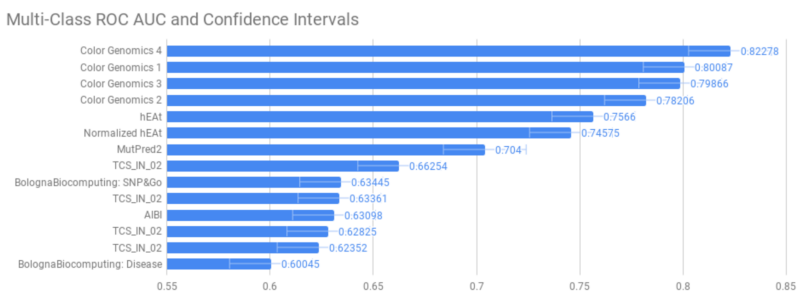
While performance is strong, the key differentiators behind LEAP’s prediction accuracy aren’t driven by model choice, but rather by the underlying data it uses. Differentiators compared to other variant predictors include:
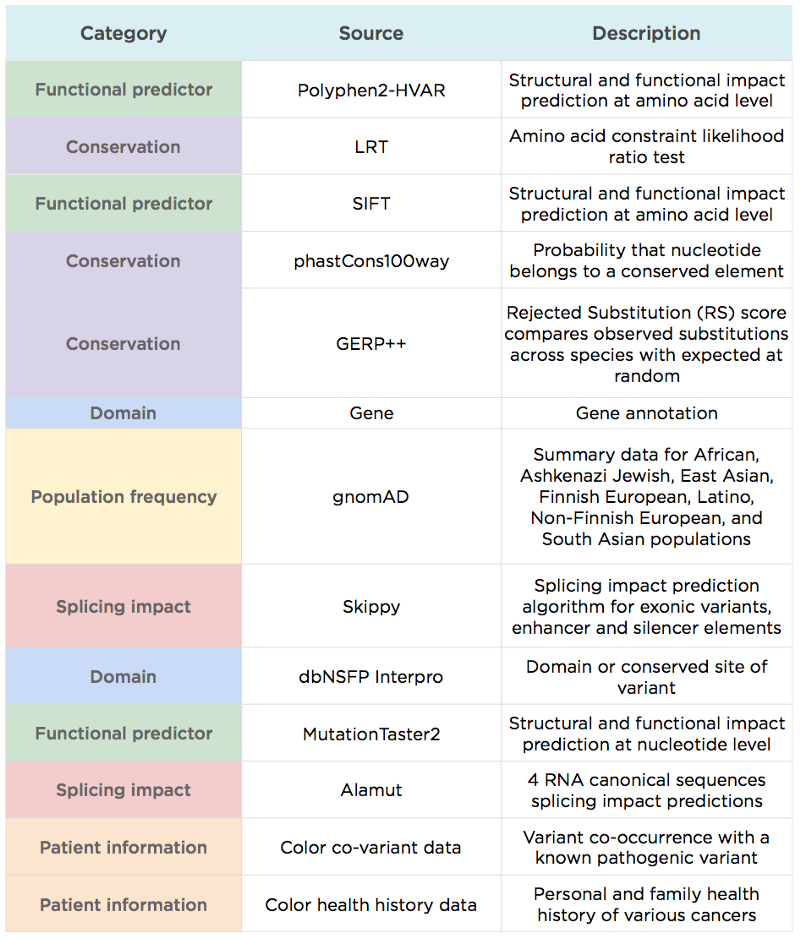
– LEAP uses additional information commonly used by variant scientists to assess pathogenicity (e.g., population frequency), in addition to information considered by most other variant predictors like REVEL (e.g., functional prediction and evolutionary conservation scores).
– LEAP takes into account information at the sample level, including phenotypic or personal and family history information, which improves both precision and recall.
– LEAP was trained on a unified cancer variant database with consistent classifications, based on ACMG guidelines and signed off by board-certified medical geneticists. One commonly cited pitfall for pathogenicity predictors is the lack of standardized classifications; many other models are trained on aggregating “consensus” classifications from public databases.
– We applied machine learning best practices to our feature processing and model validation, including scaling numeric features, binarizing categorical features, and assessing predictions on a holdout set.
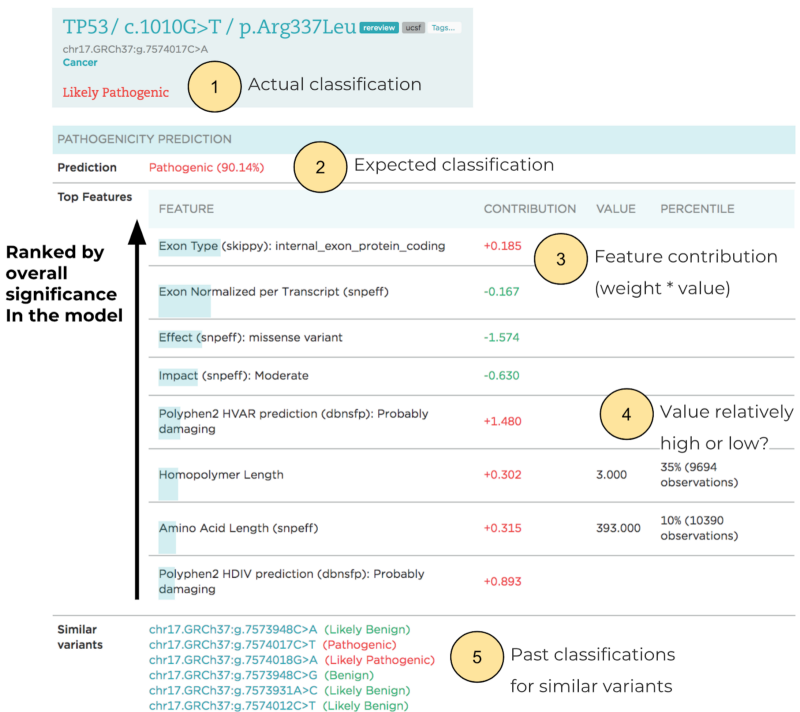
More importantly, particularly for application in a clinical setting, LEAP not only produces accurate predictions, but also explains them by providing each prediction’s rationale and driving features. The outputs are visualized for variant scientists, and highlight the contributions made by individual evidence to predictions and the overall recommendation. Details include the expected variant classification, the relative contribution of features, and classifications for variants in nearby genomic positions.
Applications in a Clinical Setting
Variant interpretation is a critical component of genetic testing, and is performed by highly-trained professionals with deep understanding of genetics. The stakes are high: incorrectly interpreting a disease-causing variant as a benign one, or vice versa, can have significant consequences for an individual. LEAP’s purpose is not to replace or automate this process, but rather to make variant interpreters more efficient.
Some concrete ways in which LEAP improves the interpretation process include:
– Prioritization: Variants that are at high likelihood of being pathogenic, or variants that are close to the decision boundary and difficult to interpret, may be flagged for expedited interpretation by geneticists, or referred to more senior ones.
– Re-interpretation: It’s common for interpretations of variants to be revisited periodically to make sure they’re up to date; at Color, non-benign variants are re-reviewed every 6 or 12 months. With LEAP, we can dynamically make this period more or less frequent, depending on the variant’s predicted pathogenicity.
– Visual aid: Finally, a variant interpreter’s workflow includes reviewing evidence from different sources (literature, known databases, and so on). LEAP summarizes much of this evidence in one place andprovides a data-driven estimate of each data point’s importance, impact, and expected overall outcome, helping the interpreter check for any discrepancies and unexpected results.
Next Steps
LEAP is a variant interpretation tool that uses machine learning to combine multiple categories of evidence, aid variant interpretation, and ultimately improve the accuracy and efficiency of our clinical reporting processes. The initial version of LEAP was trained using logistic regression, which provides visibility into individual feature contribution to a given variant pathogenicity prediction, but represents contribution in a linear fashion. A non-linear (e.g., tree-based) approach, by contrast, could capture more nuanced patterns and provide more hierarchical rationales for predictions, which may be more aligned with a variant scientist’s decision process.
Gene holdout cross-validation results also show the model’s robustness and extensibility to different loss-of-function genes for cancer. As a further extension, we’re working on applying a similar model to other health conditions, such as cardiovascular disease. These conditions are generally less well understood than cancer, but as genetics starts to play a larger role, machine learning can help us generate new variant interpretation criteria, and increase the efficiency of our variant interpretation process.



