News & Articles
The case for population screening for BRCA, Lynch syndrome, and familial hypercholesterolemia
Alicia Zhou
In 1994, Dr. Mary-Claire King discovered the BRCA1 gene. In the 24 years since that landmark discovery, the field of clinical genetics has grown by leaps and bounds. We now have a much clearer understanding of many inherited conditions, from the genes that are implicated in each condition to the prevalence of mutations across broad populations. With the price of next generation sequencing continuing to become more affordable, we are entering an era where true population screening for genetic conditions has become a reality. Indeed, newborn heel prick screening for phenylketonuria (PKU), cystic fibrosis, and congenital hypothyroidism is already common practice in most hospitals across the United States. Today, we would like to share the data in support of population screening for three genetic conditions: hereditary breast and ovarian cancer (BRCA1 and BRCA2), Lynch syndrome, and Familial Hypercholesterolemia (FH).
Our Considerations for Population Screening
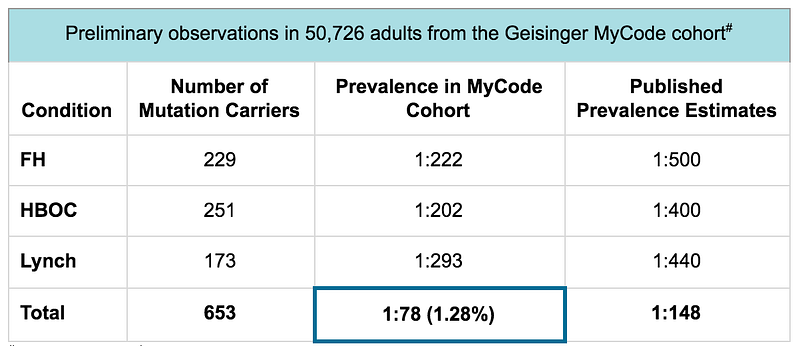
Actionable screening: These three genetic conditions were identified by the Center for Disease Control (CDC) as “Tier 1 genomic applications” because they are highly penetrant conditions with well-defined genetic causes (HBOC, Lynch, and FH) and have well-established clinical interventions to improve health and prevent disease (HBOC, Lynch, and FH). The penetrance of these three genetic conditions has been determined in population studies¹-¹⁰. Until recently, the true prevalence of gene mutations in the CDC Tier 1 genomic conditions was still unknown. However, thanks to various large-scale population health research efforts, we have come to appreciate that the prevalence is higher within the average population than previously described. In fact, data from the Geisinger MyCode program have shown that the prevalence for gene mutations in the CDC Tier 1 genomic conditions is 1:78 — double the estimated prevalence from previously reported studies.
Cost-effectiveness: To justify broad implementation, population screening must be cost-effective. By shifting spend from the treatment of medical conditions to the prevention of those conditions, costs can be decreased and life-years can be saved. For the CDC Tier 1 genomic conditions, there is a growing body of literature that supports the cost-effectiveness of population testing, including an analysis of universal BRCA1 and BRCA2 screening in JAMA Oncology¹² which stated that affordable testing for all could be cost-effective:


Broad-based patient recruitment: Broad-based access to genetic testing is critical for the implementation of population screening. Over the past three years, Color has developed an end-to-end product experience that engages and educates people about their genetic risk for inherited conditions. We have meticulously designed and created user-friendly products that make getting a genetic test simple and easy. As part of this, we have implemented easy-to-use online enrollment workflows that meet people where they are — whether at home or in-clinic — for sample collection, consent, and gathering of detailed health history information.

Genetic counselor access: Genetic counseling is an important and indispensable part of genetic testing. However, to date, access to genetic counseling has been a bottleneck to achieving population-scale genetic testing¹³. In order to make genetic counseling scaleable, we have utilized software and technology to help our genetic counselors deliver effective and efficient genetic counseling. This includes working with genetic counselors to create a series of user-friendly and easy-to-read reports; developing online scheduling tools that allow people to schedule their own appointments; and delivering counseling over the phone to decrease the barrier to access for this important service. Together, these resources allow us to improve people’s experiences while creating a new model for scaling genetic counseling to broader populations.

Cascade screening: Once mutation carriers are identified, an important next step is to test at-risk family members¹⁴. However, family testing, or cascade testing, has historically been difficult to execute. Testing is expensive, the burden of informing and educating family members falls on the mutation carrier themselves, and family members may be in geographically diverse locations. We have developed the Color Family Testing Program to overcome these traditional barriers. By bringing together a low price point for access, digital technologies that help connect family members to testing, and easier processes for participants, such as in-home sample collection, we have created a program that can increase the impact of population screening efforts in any setting.
Nearly 4–5 million people in the United States are at increased risk for hereditary breast and ovarian cancer, Lynch syndrome, and familial hypercholesterolemia, yet it’s estimated that 90% of affected individuals are unaware of their risk¹⁵. Identifying these individuals and their families through population screening can increase intervention, improve health outcomes, and shift healthcare spending from treatment to prevention.
References
- Akbari MR, Gojska N, Narod SA. Coming of age in Canada: a study of population-based genetic testing for breast and ovarian cancer. Curr Oncol. 2017 Oct;24(5):282–283. doi: 10.3747/co.24.382
- Gabai-Kapara E, et al. Population-based screening for breast and ovarian cancer risk due to BRCA1 and BRCA2. PNAS. 2014 Sep 30;111(39):14205–10. doi: 10.1073
- Metcalfe KA, et al. The risk of breast cancer in BRCA1 and BRCA2 mutation carriers without a first-degree relative with breast cancer.
Clin Genet. 2018 May;93(5):1063–1068. doi: 10.1111/cge - Kuchenbaecker KB, et al. Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA. 2017 Jun 20;317(23):2402–2416. doi: 10.1001/jama.2017.7112.
- Pearlman R, et al. Prevalence and Spectrum of Germline Cancer Susceptibility Gene Mutations Among Patients With Early-Onset Colorectal Cancer. JAMA Oncol. 2017 Apr 1;3(4):464–471. doi: 10.1001/jamaoncol.2016.5194.
- Stoffel EM, et al. Germline Genetic Features of Young Individuals With Colorectal Cancer. Gastroenterology. 2018 Mar;154(4):897–905.e1. doi: 10.1053/j.gastro.2017.11.004.
- Chen FW, et al. Low Prevalence of Criteria for Early Screening in Young-Onset Colorectal Cancer. Am J Prev Med. 2017 Dec;53(6):933–934. doi: 10.1016/j.amepre.2017.07.016.
- Akioyamen LE, et al. Estimating the prevalence of heterozygous familial hypercholesterolaemia: a systematic review and meta-analysis. BMJ Open. 2017 Sep 1;7(9):e016461. doi: 10.1136/bmjopen-2017–016461.
- Abul-Husn NS, et al. Genetic identification of familial hypercholesterolemia within a single U.S. health care system. Science. 2016 Dec 23;354(6319). pii: aaf7000. doi: 10.1126/science.aaf7000.
- Khera AV, et al. Diagnostic Yield and Clinical Utility of Sequencing Familial Hypercholesterolemia Genes in Patients With Severe Hypercholesterolemia. J Am Coll Cardiol. 2016 Jun 7;67(22):2578–89. doi: 10.1016/j.jacc.2016.03.520.
- Dewey FE, Murray MF, Overton JD, et al. Distribution and clinical impact of functional variants in 50,726 whole-exome sequences from the DiscovEHR study. Science. 2016;354(6319). doi:10.1126/science.aaf6814
12. Long EF, Ganz PA. Cost-effectiveness of Universal BRCA1/2 Screening: Evidence-Based Decision Making. JAMA Oncol. 2015;1(9):1217–1218.
13. McPherson E, Zaleski C, Benishek K, et al. Clinical genetics provider real-time workflow study. Genet Med. 2008;10(9):699–706.
14. NCCN Genetic/Familial High-Risk Assessment: Breast and Ovarian Version 2.2017. NCCN.org. https://www.nccn.org/professionals/physician_gls/pdf/genetics_screening.pdf. Published December 7, 2016. Accessed February 2017.
15. Plichta et al. What’s New in Genetic Testing for Cancer Susceptibility? Oncology Journal. 2016 Sep 15;30(9):787–99.



